According to Wikipedia, propensity score matching (PSM) is a “statistical matching technique that attempts to estimate the effect of a treatment, policy, or other intervention by accounting for the covariates that predict receiving the treatment”. In a broader sense, propensity score analysis assumes that an unbiased comparison between samples can only be made when the subjects of both samples have similar characteristics. Thus, PSM can not only be used as “an alternative method to estimate the effect of receiving treatment when random assignment of treatments to subjects is not feasible” (Thavaneswaran 2008). It can also be used for the comparison of samples in epidemiological studies. Let’s give an example:
Health-related quality of life (HRQOL) is considered an important outcome in cancer therapy. One of the most frequently used instruments to measure HRQOL in cancer patients is the core quality-of-life questionnaire of the European Organisation for Research and Treatment of Cancer. The EORTC QLQ-C30 is a 30-item instrument comprised of five functioning scales, nine symptom scales and one scale measuring Global quality of life. All scales have a score range between 0 and 100. While high scores of the symptom scales indicate a high burden of symptoms, high scores of the functioning scales and on the GQoL scale indicate better functioning resp. quality of life.
However, without having any reference point, it is difficult if not impossible to interpret the scores. Fortunately, the EORTC QLQ-C30 questionnaire was used in several general population surveys. Therefore, patient scores may be compared against scores of the general population. This makes it far easier to decide whether the burden of symptoms or functional impairments can be attributed to cancer (treatment) or not. PSM can be used to make both patient and population samples comparable by matching for relevant demographic characteristics like age and sex.
In this blog post, I show how to do PSM using R. A more comprehensive PSM guide can be found under: “A Step-by-Step Guide to Propensity Score Matching in R“.
Creating two random dataframes
Since we don’t want to use real-world data in this blog post, we need to emulate the data. This can be easily done using the Wakefield package.
In a first step, we create a dataframe named df.patients. We want the dataframe to contain specifications of age and sex for 250 patients. The patients’ age shall be between 30 and 78 years. Furthermore, 70% of patients shall be male.
set.seed(1234)
df.patients <- r_data_frame(n = 250,
age(x = 30:78,
name = 'Age'),
sex(x = c("Male", "Female"),
prob = c(0.70, 0.30),
name = "Sex"))
df.patients$Sample <- as.factor('Patients')
The summary-function returns some basic information about the dataframe created. As we can see, the mean age of the patient sample is 53.7 and roughly 70% of the patients are male (69.2%).
summary(df.patients) ## Age Sex Sample ## Min. :30.00 Male :173 Patients:250 ## 1st Qu.:42.00 Female: 77 ## Median :54.00 ## Mean :53.71 ## 3rd Qu.:66.00 ## Max. :78.00
In a second step, we create another dataframe named df.population. We want this dataframe to comprise the same variables as df.patients with different specifications. With 18 to 80, the age-range of the population shall be wider than in the patient sample and the proportion of female and male patients shall be the same.
set.seed(1234)
df.population <- r_data_frame(n = 1000,
age(x = 18:80,
name = 'Age'),
sex(x = c("Male", "Female"),
prob = c(0.50, 0.50),
name = "Sex"))
df.population$Sample <- as.factor('Population')
The following table shows the sample’s mean age (49.5 years) and the proportion of men (48.5%) and women (51.5%).
summary(df.population) ## Age Sex Sample ## Min. :18.00 Male :485 Population:1000 ## 1st Qu.:34.00 Female:515 ## Median :50.00 ## Mean :49.46 ## 3rd Qu.:65.00 ## Max. :80.00
Merging the dataframes
Before we match the samples, we need to merge both dataframes. Based on the variable Sample, we create a new variable named Group (type logic) and a further variable (Distress) containing information about the individuals’ level of distress. The Distress variable is created using the age-function of the Wakefield package. As we can see, women will have higher levels of distress.
mydata <- rbind(df.patients, df.population)
mydata$Group <- as.logical(mydata$Sample == 'Patients')
mydata$Distress <- ifelse(mydata$Sex == 'Male', age(nrow(mydata), x = 0:42, name = 'Distress'),
age(nrow(mydata), x = 15:42, name = 'Distress'))
When we compare the distribution of age and sex in both samples, we discover significant differences:
pacman::p_load(tableone)
table1 <- CreateTableOne(vars = c('Age', 'Sex', 'Distress'),
data = mydata,
factorVars = 'Sex',
strata = 'Sample')
table1 <- print(table1,
printToggle = FALSE,
noSpaces = TRUE)
kable(table1[,1:3],
align = 'c',
caption = 'Table 1: Comparison of unmatched samples')
Table 1: Comparison of unmatched samples
| Patients | Population | p | |
|---|---|---|---|
| n | 250 | 1000 | |
| Age (mean (sd)) | 53.71 (13.88) | 49.46 (18.33) | 0.001 |
| Sex = Female (%) | 77 (30.8) | 515 (51.5) | <0.001 |
| Distress (mean (sd)) | 22.86 (11.38) | 25.13 (11.11) | 0.004 |
Furthermore, the level of distress seems to be significantly higher in the population sample.
Matching the samples
Now, that we have completed preparation and inspection of data, we are going to match the two samples using the matchit-function of the MatchIt package. The method command method="nearest" specifies that the nearest neighbors method will be used. Other matching methods are exact matching, subclassification, optimal matching, genetic matching, and full matching (method = c("exact", "subclass", "optimal", ""genetic", "full")). The ratio command ratio = 1 indicates a one-to-one matching approach. With regard to our example, for each case in the patient sample exactly one case in the population sample will be matched. Please also note that the Group variable needs to be logic (TRUE vs. FALSE).
set.seed(1234) match.it <- matchit(Group ~ Age + Sex, data = mydata, method="nearest", ratio=1) a <- summary(match.it)
For further data presentation, we save the output of the summary-function into a variable named a.
After matching the samples, the size of the population sample was reduced to the size of the patient sample (n=250; see table 2).
kable(a$nn, digits = 2, align = 'c',
caption = 'Table 2: Sample sizes')
Table 2: Sample sizes
| Control | Treated | |
|---|---|---|
| All | 1000 | 250 |
| Matched | 250 | 250 |
| Unmatched | 750 | 0 |
| Discarded | 0 | 0 |
The following output shows, that the distributions of the variables Age and Sex are nearly identical after matching.
kable(a$sum.matched[c(1,2,4)], digits = 2, align = 'c',
caption = 'Table 3: Summary of balance for matched data')
Table 3: Summary of balance for matched data
| Means Treated | Means Control | Mean Diff | |
|---|---|---|---|
| distance | 0.23 | 0.23 | 0.00 |
| Age | 53.71 | 53.65 | 0.06 |
| SexMale | 0.69 | 0.69 | 0.00 |
| SexFemale | 0.31 | 0.31 | 0.00 |
The distributions of propensity scores can be visualized using the plot-function which is part of the MatchIt package .
plot(match.it, type = 'jitter', interactive = FALSE)
Here is the plot:
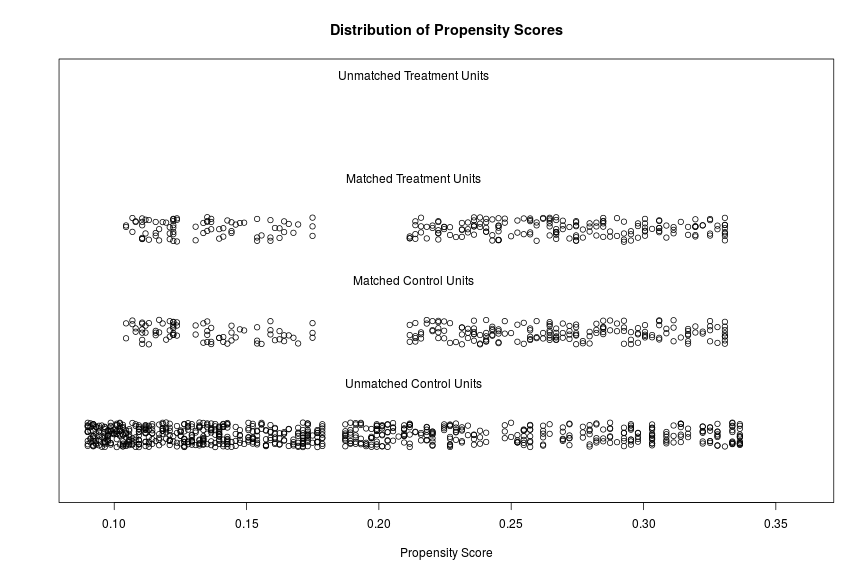
Saving the matched samples
Finally, the matched samples will be saved into a new dataframe named df.match.
df.match <- match.data(match.it)[1:ncol(mydata)] rm(df.patients, df.population)
Eventually, we can check whether the differences in the level of distress between both samples are still significant.
pacman::p_load(tableone)
table4 <- CreateTableOne(vars = c('Age', 'Sex', 'Distress'),
data = df.match,
factorVars = 'Sex',
strata = 'Sample')
table4 <- print(table4,
printToggle = FALSE,
noSpaces = TRUE)
kable(table4[,1:3],
align = 'c',
caption = 'Table 4: Comparison of matched samples')
| Patients | Population | p | |
|---|---|---|---|
| n | 250 | 250 | |
| Age (mean (sd)) | 53.71 (13.88) | 53.65 (13.86) | 0.961 |
| Sex = Female (%) | 77 (30.8) | 77 (30.8) | 1.000 |
| Distress (mean (sd)) | 22.86 (11.38) | 24.13 (11.88) | 0.222 |
With a p-value of 0.222, Student’s t-test does not indicate significant differences anymore. Thus, PSM helped to avoid an alpha mistake.
PS 1: The packages used in this blog post can be loaded/installed using the following code:
pacman::p_load(knitr, wakefield, MatchIt, tableone, captioner)
PS 2: Thanks very much to my colleague Katharina Kuba for telling me about the MatchIt package.
Greetings
I am using tableone package in r and I would like to express my appreciation to your efforts.
I have a question, after propensity score matching, I would like to compare categorical variables among both matched arms using McNemar test, is there a way to do that in tableone package instead of the regular chi square ?
Appreciate your help and time;
Mohamed
This is a brilliant tutorial. Thank you so much. I have one small question and was wondering if you advise me on the matter. I have a situation where my control is n = 300 and my treated is n = 600. After matching the samples, the size of the treated sample has now been reduced to the size of the control sample (n=300). It has done the opposite. Would this be alright to do it the other way round?
Hello
I am working on research project where I am using MatchIt package. I want a solution for this problem:
General Syntax is:
matchit(MatchLog ~ Age + Gender, data=matchframe, method=”nearest”, ratio=3)
Here Age Gender are the columns we can to match
Question: For my project I am using some algorithm to select few variables from dataframe by applying some algorithm
So I want to write the matchit function specifying the column names directly using loop without manually typing the column names like Age + Gender
I can go through your material without difficulty
Still not using in work or life
but thank you very much for sharing
Hello! I’m trying to construct a data frame of controls matched by age and gender, and I’m getting the following error code:
> matchit(MatchLog ~ Age + Gender, data=matchframe, method=”nearest”, ratio=3)
Error in matchit(MatchLog ~ Age + Gender, data = matchframe, method = “nearest”, :
Missing values exist in the data
I used the is.na function to check all variables in my dataset, and found no cells with N/A as their value. Is there another way that there may be ‘missing data’ in my dataset?
The MatchLog variable is formatted as logical.
Thanks so much!
Figured out a workaround. Made a separate data frame with just the variables I needed for matching, plus a column with an identifying number. Used the smaller data frame to make matches, then re-joined the data afterwards. Hope this helps someone out!
hello I am trying to psm by R with matchit.
It seems that every time I do matching the results are different.
Is it normal that way?
Hello! When you use Nearest Neighbourhood with a defined caliper value (maximum propensity score difference) without setting a seed for the random process, than you will always get different results! By setting a seed, you should get the same results or without the usage of a caliper, you should also always get the same results!
Thanks a lot for information. It is just that… I am not very familiar with R and have no idea what is “setting a seed”.
is there a code for it???
You are welcome! Yes, ‘set.seed(1234)’ should do the trick, as you can see in the code windows of the article of Norbert Köhler above…! Remember, as long as you use a caliper the controls that fall within the caliper range of a treated are chosen randomly and that’s why you get different results (correct habit) without setting a seed for the random process. Note: when you do not use a caliper but choose ‘random’ for the matching order (despite ‘largest to smallest’ or ‘samllest to largest’), i.e. before the matching starts the treated and controls are ordered randomly, you invoke again a random process that will lead to different results without setting a seed…
I got it. I was wondering if process itself was coded wrong. You are such a big help thanks a lot-!!! It is just that……
what if the p value for outcome result differ time to time when I match?? Is it statisticily alright when I use the set which shows the result I want? or is there other ways to confirm the “right” matching??
You are welcome!
There is no ‘right’ or ‘wrong’ matching but matching of ‘good’ or ‘bad’ quality, determined by the balance checks of the ‘MatchIt’ package (have a look at Ho, Imai, King & Stuart onhttps://www.jstatsoft.org/article/view/v042i08), to reduce the effects of confounding (have also a look at Austin 2011 on https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3144483/). Give it a try!
wow thanks so much.It really helped a lot. now I set seed everytime I do matching.
You are welcome! I tried to answer you two times, but my posts do not seem to be approved by ‘disqus’…!? I’m sorry…!
Instead of ‘wrong’ and ‘right’ matching try to think of matching of ‘good’ and ‘bad’ quality, determined by the balance checks of the ‘MatchIt’ package (details in Ho, Imai, King & Stuart on https://www.jstatsoft.org/article/view/v042i08), to reduce the effects of confounding (see also Austin on https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3144483/). When you have achieved matching results of sufficient quality, these should lead to robust results for your modeling choices in your observational study…!
Hi,
Excellent tutorial! Thank you
However, I cannot create table1 with this code:
table1 <- CreateTableOne(vars = c('Age', 'Sex', 'Distress'),
data = mydata,
factorVars = 'Sex',
strata = 'Sample')
This is the error I get:
None of the stratifying variables have 2+ valid levels.
Thanks
Thanks a lot! Have you created the dataframe ‘mydata’ in advance? You may download my .Rmd file under the following link: https://drive.google.com/open?id=0B61FC2RGlvl3eWY0SVpjOVNvVW8
Does it work on your machine?
Hi Norbert,
Thank you. The problem was that I used this incorrectly:
pacman::p_load(tableone). I did not know what :: meant 🙁
It works now. I believe for many users of your tutorials it would be very helpful if you could comment briefly on the pacakges you use in your scritps. All of them are very useful but I never heard about them…
Thanks very much for your advice! I will do so in my future posts. 🙂